|
miRvestigator Framework Help
|
|
What is the miRvestigator Framework?
|
The miRvestigator framework is designed to take as input a list of co-expressed genes and will return the most likely miRNA regulating these genes. It does this by searching for over-represented sequence motifs in the 3' untranslated regions (UTRs) of the genes using Weeder and then comparing this to the miRNA seed sequences in miRBase using our custom built miRvestigator hidden Markov model (HMM).
|
|
|
Entering a Co-Expressed Gene List
|
|
What is gene Co-Expression?
Post-transcriptional regulation by a miRNA is mediated through binding to complementary sequences in the 3' untranslated regions (UTRs) of transcripts and generates a biological context-specific signature of down-regulation for these transcripts, which is called a co-expression signature. Co-expression signatures observed in transcriptome studies are thus assumed to be of biological origins whereby the expression of genes are regulated by shared factors (transcription factors, miRNAs, genetics or environmental factors). There are many methods available to identify co-expression signatures, and the miRvestigator framework does not bias toward any particular method.
Do I need quantitative expression data to use miRvestigator?
The input for miRvestigator is a gene list (e.g. 79073, 900, 29957, 11167, 4154, 84270, 84061, 4001, 26503, 4086, 51585, 4734, 5873) and thus does not require quantitative expression data as input. However, it is assumed that some other information was integrated to identify that set of genes, such as gene co-expression, specific miRNA perturbation, etc.
What species are supported by miRvestigator?
miRvestigator currently supports:
If the species you study has miRNAs and is not on this list please e-mail us and we will do our best to add your species of interest to miRvestigator.
Can miRvestigator include viral miRNAs?
For those who study the effects of viral miRNAs on host gene expression we have added the ability to include viral miRNAs. By selecting the Yes radio button the viral miRNAs will be searched along with the host species miRNAs. Note that perfectly overlapping seed seqeunces of a viral miRNA with a host miRNA will be concatenated as we cannot separate these influences. (NOTE: All viral miRNAs are included so you will have to filter them for those that you are interested in, please consult miRBase for the species codes.)
What if I don't have one of your supported gene identifiers (IDs)?
Currently the miRvestigator framework is able to take Entrez gene IDs, Ensembl gene IDs, RefSeq transcript IDs and official gene symbols as input. To convert any other type of identifier to Entrez gene IDs please use the DAVID bioinformatics resource gene ID conversion tool. The DAVID gene ID conversion tool has a great help page please visit it if you have any questions.
What types of delimiters can be entered into the form?
You can enter the gene IDs separated by commas, spaces, tabs or newlines.
Loading and Using Sample Data Load the data by simply clicking the button with the corresponding miRNA on it, which will load the data and setup any parameters. Then click the submit button at the bottom of the page.
Where does the sample data come from?For the H. sapiens (human) sample data are co-expression signatures reduced to Entrez ID gene lists from studies where a miRNA was experimentally perturbed and the resultant effect ascertained by transcriptome measurements (e.g. gene expression microarray). To provide a working sample dataset for other species where an experimentally perturbed datasets were not yet available we utilized TargetScan miRNA target mRNA predictions for miR-1.
- C. elegans
- D. melanogaster
- G. gallus
- H. sapiens
- hsa-miR-1 - The HEK293T human embryonic kidney cell line with FLAG-Ago2 was transfected with and without exogenous miRNA. Genes significantly enriched (1% local FDR) in the co-immunoprecipitates of the FLAG-Ago2 transfected cell lines where the miRNA was included were considered targets of the miRNA.
Hendrickson, D.G., Hogan, D.J., Herschlag, D., Ferrell, J.E. & Brown, P.O. Systematic identification of mRNAs recruited to argonaute 2 by specific microRNAs and corresponding changes in transcript abundance. PLoS ONE 3, e2126 (2008).
- hsa-miR-9 - Only genes exceeding median expression in at least half of the experiments were assayed for differential expression. Differential expression (P-value < 0.001) was required to be consistently observed at both the 12 hour and 24 hour time points after miRNA transfection in HeLa cells. This dataset was reanalyzed from data in GEO data repository GSE8501 using the specification in the publication stated above to identify perturbed miRNA target genes.
Grimson, A. et al. MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol. Cell 27, 91-105 (2007).
- hsa-miR-16 - Down-regulated genes were identified at 24 hours after miRNA transfection with consistent differential expression (P-value < 0.01) in HCT116 and DLD-1 colon tumor cell lines hypomorphic for Dicer (HCT116 Dicerex5 and DLD-1 Dicerex5, respectively) 2. Consensus miRNA down-regulated transcripts were also required to be significantly down-regulated (P-value < 0.05) at 6 hours post-transfection in the colon cancer HTC116 Dicerex5 cell line 2.
Linsley, P.S. et al. Transcripts targeted by the microRNA-16 family cooperatively regulate cell cycle progression. Mol. Cell. Biol 27, 2240-2252 (2007).
- hsa-miR-132 - Only genes exceeding median expression in at least half of the experiments were assayed for differential expression. Differential expression (P-value < 0.001) was required to be consistently observed at both the 12 hour and 24 hour time points after miRNA transfection in HeLa cells. This dataset was reanalyzed from data in GEO data repository GSE8501 using the specification in the publication stated above to identify perturbed miRNA target genes.
Grimson, A. et al. MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol. Cell 27, 91-105 (2007).
- hsa-miR-142-3p - Only genes exceeding median expression in at least half of the experiments were assayed for differential expression. Differential expression (P-value < 0.001) was required to be consistently observed at both the 12 hour and 24 hour time points after miRNA transfection in HeLa cells. This dataset was reanalyzed from data in GEO data repository GSE8501 using the specification in the publication stated above to identify perturbed miRNA target genes.
Grimson, A. et al. MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol. Cell 27, 91-105 (2007).
- hsa-miR-184 - Reported consistent down-regulation (log(FC) < -1, P-value < 0.05) of non-redundant genes in the glioma cell lines A172 and T98G 72 hours after miRNA transfection that also were predicted to be targets of this same miRNA by miRBase target database.
Malzkorn, B. et al. Identification and functional characterization of microRNAs involved in the malignant progression of gliomas. Brain Pathol 20, 539-550 (2010).
- M. musculus
- mmu-miR-1 - TargetScan 5.1 mmu-miR-1 target gene predictions.
Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets.Cell 120(1):15-20 (2005).
- mmu-miR-122 - Experimentally derived mmu-miR-122 target genes using antagomirs.
Kr�tzfeldt J, Rajewsky N, Braich R, Rajeev KG, Tuschl T, Manoharan M, Stoffel M. Silencing of microRNAs in vivo with 'antagomirs'. Nature. 2005 Dec 1;438(7068):685-9.
- R. norvegicus
- H. sapiens infected with Kaposi-Sarcoma-Associated Herpes Virus
|
|
|
|
Parameters for Weeder
|
|
What is Weeder?
Weeder is an enumerative algorithm used to identify over-represented sequence motifs in the miRvestigator framework.
What are the motif sizes?
Weeder identifies both 6 base-pair (bp) and 8bp motifs for each run. Using this option it is possible to choose which motif size is subsequently used in the miRvestigator HMM is set by this option. Picking only one of the motif sizes will make the run faster. By default we have chosen to use 8bp motifs. (Note: It is possible that the 6bp motif and the 8bp motif will be different.)
What background model does Weeder use?
Weeder uses the background model to determine whether or not an oligo is enriched above background. Each different species has a different background model based upon the sequence composition of the specified region in that species. The Weeder documentation states that the "Default Weeder Model" should be sufficient for 3' UTRs even though it was built based up on sequences upstream from the start codon, which was consistent with our findings. Although in certain cases a model built upon the annotated 3' UTR sequences from UCSC was helpful. Therefore, by default the "Default Weeder Model" is chosen although it may be best to try the "3' UTR Specific Model" as well. (Note: It is likely that the 3' UTR background model would improve given more complete 3'UTR annotations.)
What target site quality threshold is best?
The way Weeder is setup to run in the miRvestigator framework it identifies motifs based upon allowing at max 2bp of non-complementarity. The canonical view in the field of miRNAs is that 7mer and 8mer of complementarity to the seed sequence are the most efficacious. If the target site quality threshold is set at 100% Identity then only perfect 8mer or 6mer of complementarity (depending on the motif size) are returned, and if the target site quality threshold is set at 95% identity this typically retrieves putative sites with 1bp non-complementarity to the motif. Therefore to be consistent with the canonical view of the activity of miRNAs we set the target site quality threshold by default to 95% identity. (Note: When using 6bp motifs the target quality threshold ought be set to 100% identity, and the efficacy of the 6bp predicted binding sites would need to be extensively tested experimentally.)
|
|
|
|
Parameters for miRvestigator HMM
|
|
What is the miRvestigator hidden Markov model (HMM)?
The miRvestigator hidden Markov model (HMM) is a method designed to take an over-represented sequence motif (in this case from Weeder) and compare it to all the miRNA seed sequences from miRBase using the Viterbi algorithm. The over-represented sequence motif is turned into a profile HMM and each seed sequence in miRBase is aligned to identify the best complementarity and a probability of complementarity computed using the Viterbi algorithm. Then a Complementarity p-value is calculated for each miRNA by comparing it to an exhaustive distribution of Viterbi probabilities.
What are the seed models?
Base-pairing models for the seed regions of a miRNA to the 3' UTR of target transcripts. The 8mer, 7mer-m8, and 7mer-a1 models are the canonical models of miRNA to mRNA base-pairing. The 6mer models are considered marginal models as they typically have a reduced efficacy and are more likely to occur by chance alone. By default all of the seed models are used. The seed models are described in this figure: 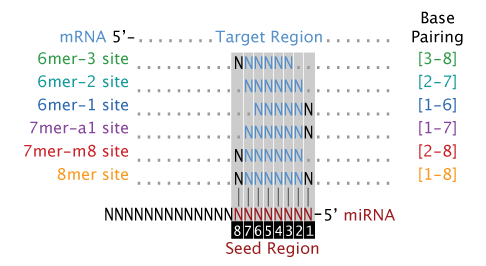 Model Wobble Base-Pairing?
Model Wobble Base-Pairing?
The miRvestigator HMM can also model G:U wobble base-pairing which has been observed in miRNA to target transcript 3' UTR complementarity. The sequence motif is unlikely to be comprised completely of instances where a G:U wobble was used in the exact same spot. However, it may be possible and we do not want to exclude the possibility. Thus the miRvestigator framework can be enabled to model these G:U base pairings once the G or U nucleotide frequency in the sequence motif column passes a specified threshold. This threshold can be set with the "Min. Freq. of G or U". By default the wobble base-pairing is not modeled but if wobble base-pairing is enabled we recommend a threshold of 0.25 for the "Min. Freq. of G or U".
|
|
|
|
Submitting a Job
|
|
How many top miRNAs to return?
miRvestigator provides as output a list of miRNAs from miRBase that are sorted based on the complementarity between their seed sequence and the motif identified from the 3' UTRs of the input sequences. We save all the results out, but allow the user to decide how many to return: Top 10, Top 25, Top 50, Top 75, Top 100 or All.
Notification via e-mail when job is finished? (Optional)
miRvestigator allows users to be notified by e-mail when there job has finished processing. Included in the e-mail will be a link to the results page. Allowing users to submit multiple jobs and access the results at their leisure. Alternatively you can bookmark either the status page or results page for your run to return to collect your results. (NOTE: If you don't receive e-mails please check your e-mail address, spam filter and add mirvestigator@systemsbiology.org to your address book. Typically by adding an e-mail address to your address book will allow e-mails from that address to bypass the spam filter.)
|
|
|
Retrieving Results
|
|
What happens after I click submit?
After clicking submit you will be taken to a status page (see Example Status Page below) which can be bookmarked to return and collect results at a later time. As your miRvestigator job is processed the status page will be updated with information about the run. The submission parameters are listed below the updates.
Example Status Page: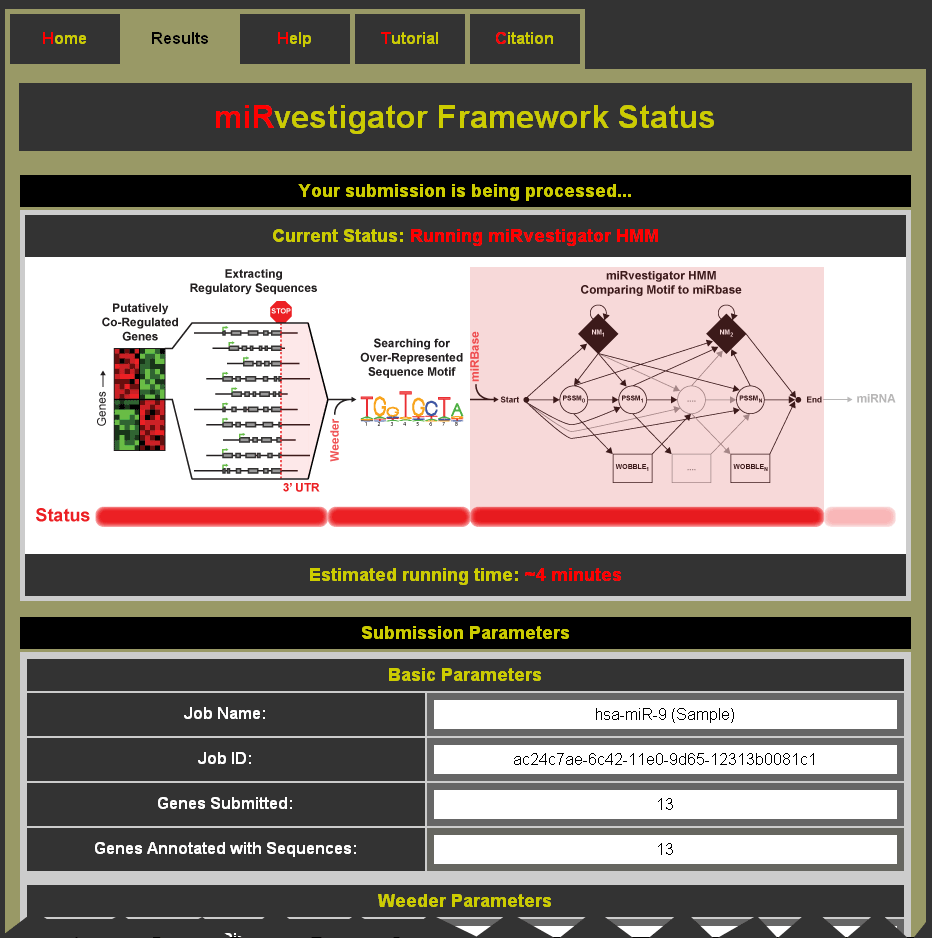
After the job has finished processing you will be taken automatically to the results page (seed Example Results Page Below) with the results for your run. The results page can also be bookmarked so that you can return to view the results at a later time. Results will be kept for at least two months.
Example Results Page: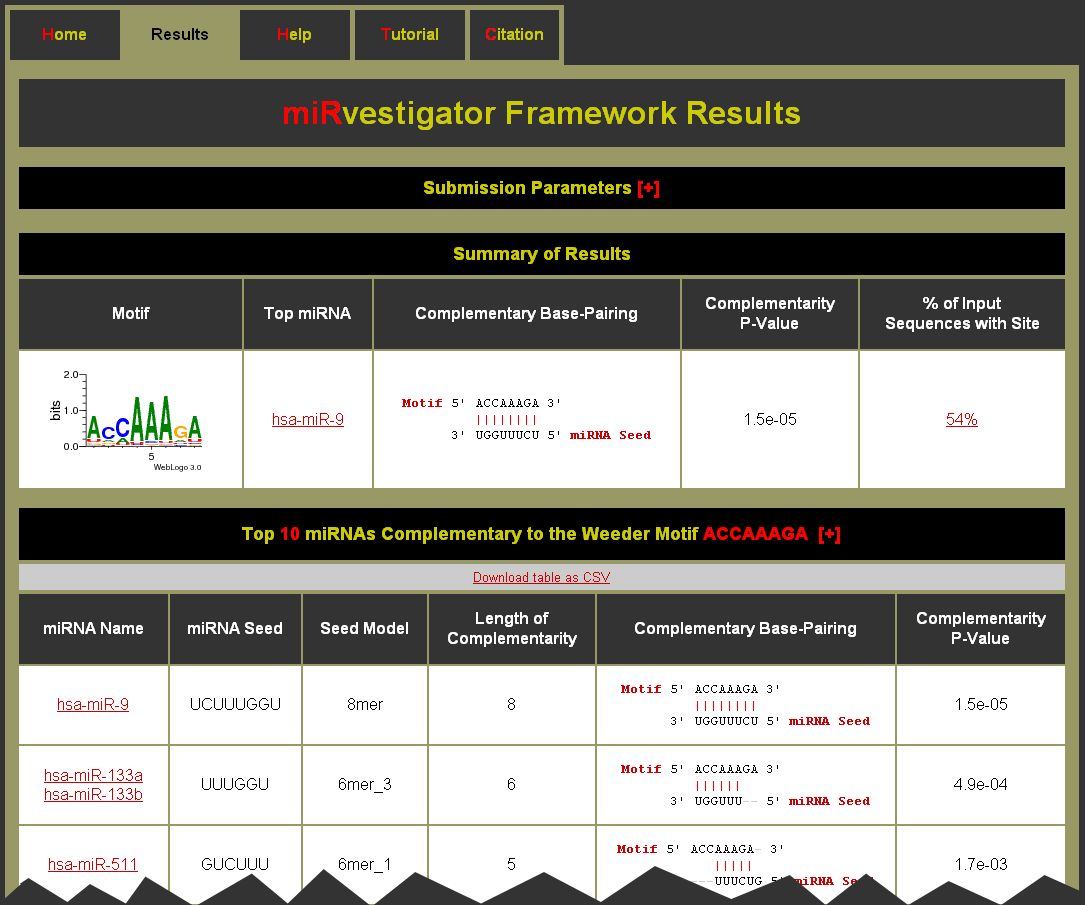 Do I have to stay on the status page in order to see the results?
Do I have to stay on the status page in order to see the results?
No you do not. Either by bookmarking the status page or by giving your e-mail address you can easily retrieve your results at a later time. (NOTE: If you don't receive e-mails please check your e-mail address, spam filter and add mirvestigator@systemsbiology.org to your address book. Typically by adding an e-mail address to your address book will allow e-mails from that address to bypass the spam filter.)
Can I retrieve my results with just the job id?
The job ID is a long character string that is unique for each job. If you retain this you can click on the results link at the top of any page (except the results page) to access the results from a previous run.
|
|
|
miRvestigator Results
|
|
Example Results Page:
What is on the results page?
The results page has at the top a hidden table of the Submission Parameters, which can be expanded by clicking on the [+]. Next is a Summary of the Results for each motif, which contains a table that has key information about each motif. For each motif we display:
- Motif - a logo plot for the motif identified from the 3' UTR sequences of the input sequences. (Note: This is also a link to the table of complementary miRNAs.)
- Top miRNA - the top complementary miRNA as determined by the miRvestigator HMM. This is a link to the miRNA in miRBase.
- Complementary Base-Pairing - complementarity of the consensus motif to the miRNA seed sequence for the top complementary miRNA.
- Complementarity P-Value - significance of the complementarity between the over-represented sequence motif and the miRNA seed sequence. (Note: A perfectly complementary 8mer seed model is 1.5e-05, for a 7mer seed model 6.1e-05, and for a 6mer seed model 0.00024.)
- % of Input Sequences with Site - percent of input sequences that had a predicted binding site passing the target site quality threshold. (Note: This is a link to the table of motif sites.)
Following the Summary of Results are the Motif Complementary miRNAs and Motif Sites tables. These two tables contain the results for the miRvestigator framework run. We describe both of these tables in detail in the next two sections.
|
|
|
miRvestigator Results: Motif Complementary miRNAs Table
|
Example Motif miRNA Complementarity Table: Can this table be downloaded?
Can this table be downloaded?
There is a download table as CSV link at the top of the complementary miRNAs table. Click on the link and open the CSV downloaded to your computer using your favorite spreadsheet application.
What do the columns mean?
- miRNA Name = The name of the name(s) for the unique seed sequence. There may be more than one miRNA annotated for a unique seed sequence because they vary in the 3' terminus of the mature miRNA. Each miRNA is a link to it's entry on miRBase
- miRNA Seed = The sequence of the most complementary seed for the over-represented motif. The seed will be as long as the seed model described in the next column.
- Seed Model = Base-pairing models for the seed regions of a miRNA to the 3' UTR of target transcripts. The 8mer, 7mer-m8, and 7mer-a1 models are the canonical models of miRNA to mRNA base-pairing. The 6mer models are considered marginal models as they typically have a reduced efficacy and are more likely to occur by chance alone. By default all of the seed models are used. The seed models are described in this figure:
- Length of Complementarity = The length of complementarity (or wobble if enabled) base-pairing between the sequence motif and the miRNA seed sequence.
- Complementary Base-Pairing = The complementarity of the over-represented sequence motif on top 5'⇒3' to the miRNA seed sequence given the seed model 3'⇒5'. (Note: | = a complementary, : = a wobble, " " (space) = not complementary, and for the sequences - = a gapping at the start or end.)
- Complementarity P-Value = Significance of complementarity between the over-represented sequence motif and the miRNA seed sequence. (Note: A perfectly complementary 8mer seed model has a Complementarity p-value of 1.5e-05, for a 7mer seed model 6.1e-05, and for a 6mer seed model 2.4e-04.)
What is considered good complementarity? This will depend upon your data and what downstream analysis you plan to do with it. But a good rule of thumb is that if you find perfect complementarity for a 7mer or 8mer (Complementarity P-Value = 6.1e-05 and 1.5e-05; respectively) this is likely to be of interest. Follow up with experimental studies will help to determine the false discovery rate for your dataset.
|
|
|
miRvestigator Results: Motif Sites Table
|
Example Motif Sites Table: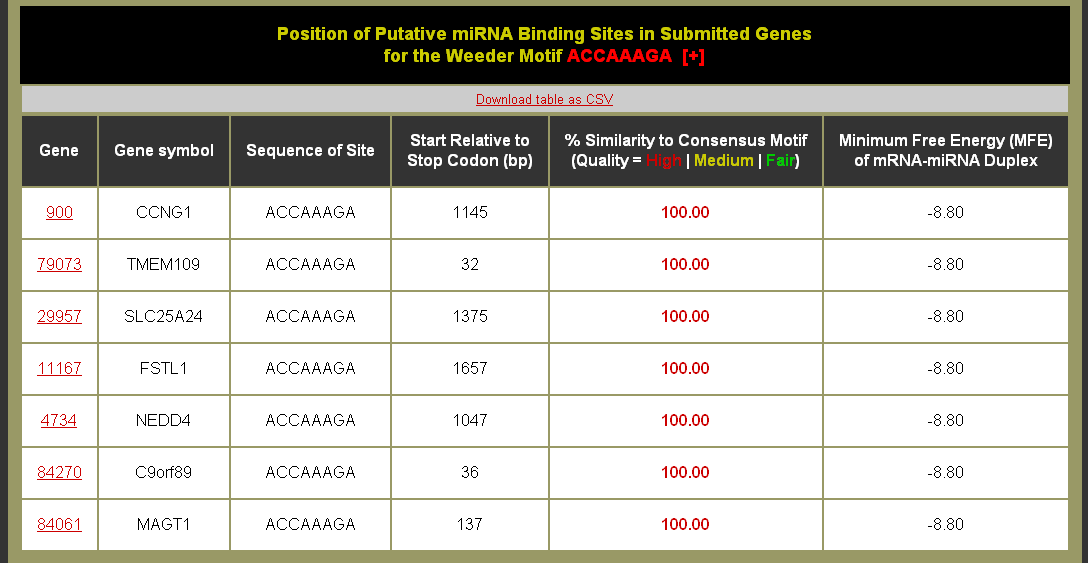 Can this table be downloaded?
Can this table be downloaded?
There is a download table as CSV link at the top of the motif sites table. Click on the link and open the CSV downloaded to your computer using your favorite spreadsheet application. (Note: The downloaded table will not be filtered by target site quality threshold.
Where do these sites come from?
As part of the miRvestigator framework Weeder provides predicted miRNA binging sites in the 3' untranslated regions (UTRs) of the analyzed genes. Predicted binding sites were split into three different similarity bins: High quality - ≥ 95% similarity to the miRNA seed sequence (red), Medium quality 95% ≥ similarity ≥ 90% to the miRNA seed sequence (yellow), and Fair quality 90% ≥ similarity ≥ 85% to the miRNA seed sequence (green). These sites can be used to develop follow-up experiments such as luciferase reporter assays to validate the efficacy of these sites.
Are these sites filtered?
As part of the parameters for the run you can set the target site quality threshold to filter for perfect complementarity (100% Identity), perfect complementarity and 1bp discrepancies (95% Identity), or down to 2bp discrepancies. We recommend sticking with 100% or 95% identity thresholds as these are the most similar to the canonical view of the mechanistic action of miRNA binding through the seed sequence with 7bp or 8bp of similarity between the miRNA seed sequence and the target mRNA. If using a 6bp motif only use 100% identity threshold, and follow these up with extensive experimental validation.
What do the columns mean?
- Gene = gene identifier from the input set that links to the mapped Entrez identifier page on NCBI.
- Gene Symbol = Official gene symbol if available.
- Site = The sequence for site identified by Weeder. If it is in square brackets indicates that the site is of lower similarity.
- Start Relative to Stop Codon = The 3' UTR begins following the stop codon (which is set at 0 base-pairs (bp)). Thus the values in this column describe the start of the site in bp after the stop codon.
- % Similarity to Consensus Motif = The similarity of the predicted site to the consensus motif is computed as a percentage. Predicted binding sites were split into three different similarity to consensus bins: High quality - ≥ 95% similarity to the miRNA seed sequence (red), Medium quality 95% ≥ similarity ≥ 90% to the miRNA seed sequence (yellow), and Fair quality 90% ≥ similarity ≥ 85% to the miRNA seed sequence (green).
- Minimum Free Energy (MFE) of mRNA-miRNA Duplex = the minimum free energy (MFE) of duplexing for the reverse complement of the motif consensus sequence and putative target site sequences using the ViennaRNA package.
|
|
|
Other Implementations of miRvestigator
|
|
Stand-alone miRvestigator for High-Throughput Screening of Motifs
This is a command-line program to identify miRNAs from a Position Specific Scoring Matrix. A PSSM is read from a user specified file and is then passed to miRvestigator which downloads the mature miRNA seed sequences from miRbase.org and compares these mature seed sequences against the PSSM. This is accomplished by converting the PSSM into a Hidden Markov Model (HMM), very similar to a profile HMM, and then using the Viterbi algorithm to simultaneously align and calculate a probability for the alignment of the miRNA seed to the HMM. A p-value is calculated by exhaustively comparing all potential miRNA seed sequences that could bind to 3' UTRs, and using simulations the p-value for the Viterbi probability is the optimal metric for gauging miRNA seed to PSSM similarity.
|
|